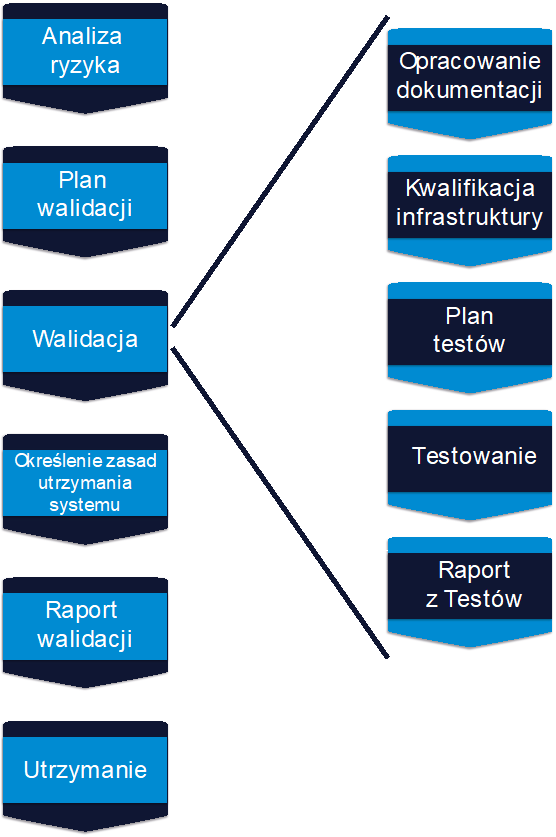
Computer systems validation is a procedure performed in every pharmaceutical or medical device company that uses IT systems at the stage of production, logistics or distribution of its products. Creating a new system and its modifications or implementing existing software requires proper project management. There are two approaches to this type of process prevalent in IT projects, namely the traditional and agile management. This article looks at the differences between the two methodologies and their advantages from the validation point of view.
The main issue is an approach to conducting projects involving the validation of computerized systems in the pharmaceutical sector. Due to serialization requirements, many pharmaceutical firms will be bound to implement some completely new software. Projects are usually delivered using either a classic waterfall or agile methodology. The question is, what are the differences between those two and when do they prove to be more effective?
With traditional methodologies, such as Prince2 or PMI, we divide a project into individual stages (analysis, design, execution and implementation). Since each phase must be completed before the next one begins, they are often called cascade projects. The project is executed according to a precise scheme; its functionality and requirements are well-known and all its areas, such as its duration, measurable business product, resources, organizational structure, including roles and responsibilities necessary for project management, are precisely defined. The project is delivered as a whole. This methodology is characterized by a strict sequence of stages. If the preceding phase has not been satisfactorily completed, the next one cannot begin. It is necessary to go back to the previous stage and make certain modifications to achieve the expected result.
In contrast, in the agile methodology such as Scrum, the plan is also defined, but everything else is flexible. The team itself is responsible for organizing and assigning tasks, and the project is completed stage by stage. Throughout the project, new needs may arise that the team can follow. The scope must be defined both in the agile and cascade projects and it may be identical, but when there is a need to modify the project or the schedule, the difference between the methodologies is clear. In agile methodologies, we have instruments that allow us to modify this scope quickly and easily. In the classic approach, we also have such procedures at hand, but they are time-consuming and require much more commitment. It is also worth mentioning that implementing a project using the waterfall approach requires more time, which usually leads to higher costs. This also entails the necessity to repeat individual stages when there are no effects or when the effects are not the ones that were expected, which, consequently, will lead to downtime.
Another methodology that should be mentioned here is the hybrid agile project management, which combines control mechanisms known from traditional methodologies, such aspects as risks, budget, time and quality, but the paradigm is the continuous collection and discovery of new requirements and following the change.
How do these differences reflect on project validation processes?
Let us agree that validation is a rather rigid method, which requires certain procedures and instructions and is nowhere near flexibility, which probably in many projects would be helpful. We can say that it is a typical cascade process and from our perspective, it is this constant variability in agile methodology that is a challenge, especially from the point of view of documentation and testing. In agile projects, it is both important and problematic to set up our validation rules to avoid too much “distortion” of the agile approach. That means we need to leave the possibility of flexible defining or changing the scope of the project, but on the other hand, we have to observe the formalities required in the validation process, namely to develop a risk analysis and to supervise the risk in the process, to plan the scope of validation work and the scope of tests, to define and develop the required technical documentation without the risk that something has been omitted or, for example, that the scope of tests has been inadequate.
You should also note that there are so-called hard-gates in the validation process that must be met before moving on to the next stage. For example, specification documentation (URS, FS, DS) must be developed before testing can begin. For instance, in agile methodologies, the dynamics of changing functionalities directly affects the documentation, which in turn translates into testing. From my perspective, the whole art of validating agile projects lies in building such an approach or validation model so that completing the formalities one way or another does not block the flexibility of changes in the project.
If the project is too simple to be broken down into smaller phases, i.e. so-called sprints in Scrum nomenclature, it is hard to talk about an agile project. Breaking a project into sprints means gradually adding functionality, which is closer to the client’s requirements from sprint to sprint. In the waterfall project we plan everything from A to Z (first of all, what the final product should look like), we go through the various stages of production and we deliver and check the final product right away, while in agile methodologies we deliver individual elements or functionalities that make up the whole. It is wrong to assume that in agile methodology the plan is improvised, because generally each action is defined and contrary to what it may seem, some agile methodologies also make use of deadlines, i.e. we have the so-called time boxes. In the Scrum methodology, each phase of work, however small, such as an iteration or a sprint, is rigidly defined. Tasks are performed precisely and all our results are verified on an ongoing basis. It’s certainly easier to verify the performance and results after each sprint because if you identify bugs in the delivered functionality from the last sprint, you can implement an improvement after the bug was identified, or schedule a fix for the next sprint. From a software development point of view, for example, this is a very big advantage. From a validation point of view, it complicates the whole situation.
Does this mean that within an iteration or a sprint, validation tasks can be carried out alongside the agile process of the whole project?
It depends on what phase the project is in. It often happens that these initial phases, when, for example, the environment is being prepared and the backend and configuration code are being created, do not contribute much to the whole and from the validator’s point of view can be lost. I have come across two approaches in this situation. In the first one, the validator is involved in the project from the beginning and starts his activities in 2/3, 3/4 of the sprint, for example checking the documentation or validating the application code. He stays on top of it and gives feedback, comments or what has been done wrong from a validation point of view. The validator has control over the entire development process on an ongoing basis and can verify individual stages or functionalities. The risk that the final product will be incompatible with business objectives and requirements and the whole process has not been conducted by the rules, regulations or good manufacturing practice is minimized. Of course, there are also disadvantages of that solution, for instance certain functionality, which was created during the sprints, is often absent in the final product because it has been abandoned. That is contrary to the validator’s approach because his role is to ensure that the solution is consistent with the business goals or specifications. In the second approach, the validator gets involved with the project in its final stages, let’s assume it to be 80% of the delivered functionality. He starts validating and working with the person responsible for the documentation, for the source code, and gets the documents that are needed at the end of the project.
The latter approach obviously has a very serious con. If everything has been done correctly, nothing is missing from the documentation, there are no mistakes and no defects, then the validation process goes smoothly. But if there are complications, deficiencies or defects, validation takes longer and has directly affected the delivery of the final validated product. A far better model is to involve the validator in the project from the very first stages, albeit to a lesser extent.
So, when should a validator or a team of validators start their work in the case of software development or implementation of the project in a pharmaceutical company?
The later we get involved in the project, the worse. The worst option is to include validation right before the tests themselves because in practice this means the most work for everyone. On the one hand, for a validation project to start, a document such as risk analysis or a validation plan has to be created. Even if we are talking about the agile methodology, or Scrum to be precise, we should get involved at the very beginning, with the risk analysis. The validator should have a say in developing such things as the test strategy, when the tests are performed, what tests are performed, what documentation is created during the project, whether it is kept meticulously, what the working environments are, what the implementation should look like, in what environments, and much more. Even if most of the validator’s job is taken into account in the testing and pre-implementation phase, our involvement in the early phases of the project will be helpful. Joining the project, later on, may lead to delays or entail a lot of additional work and costs that will be necessary to make the project compliant and meet regulations, of course when we talk about the need for validation. I am sure it is no secret to anyone that it costs the least, both in terms of time and commitment, as well as in financial terms, to amend a project at the beginning.
Including validation at the very beginning is crucial, because the project team should know what is going on and what is expected. The mere fact that there is no scope or plan of the project yet, is not a matter of agile methodology, it is a result of bad project organization. It is sometimes misunderstood in the case of IT projects that if the project is carried out using agile methodology, certain issues can be approached more freely, e.g. the documentation will be written later or plans will be made later. This is not the case. The scope of the project and its implementation plan must always be known, documented and written down. The documentation must also be provided. As I said at the beginning, the only difference is whether we can break it down into phases and whether we have procedures or functions that allow us to make these modifications.
Let’s assume that a project is being implemented in the agile methodology, but during one of the stages, a defect or non-compliance with the documentation appears. What happens then? Is the sprint or phase terminated when the functionality works? Is there any procedure for that?
We should refer here to the Scrum methodology. Sprints are usually two-week stages, during which specific tasks are planned. After each sprint, you have to present the effects, successfully complete tasks and identified errors (which need to be corrected). After the progress of the project is presented, another meeting is held with the business department to plan the scope of the next sprint. At this meeting, detailed requirements can be discussed, and any doubts as to how the requirements will be implemented can be resolved. If there are bugs, fixing them is included in the plan. In Scrum projects, no sprints are planned from the beginning to the end of the project. To put it simply, a Scrum project is a set of individual tasks that must be completed throughout the project for the final product to be formed as intended. However, the number of sprints often increases during the project because of the bugs that need to be resolved or new features that need to be added. During the sprints, certain tests in the project should also be scheduled, so that it is possible later on to prepare the documentation,, report the progress and the detected bugs, and plan to correct them.
This is also the difference between waterfall and agile methodologies. In traditional methodologies, the occurrence of a critical problem stops the project, because you have to do the tests before you can implement the whole project, so no project may begin until the problem is solved. In Agile, this doesn’t necessarily mean that the project will end up at a standstill. If a given sprint or iteration doesn’t affect other phases or functionalities (and it often doesn’t, because the next stages are only being planned), then one team can work on solving the problem, while the other one carries out other tasks. But in principle, we know about problems or bugs on an ongoing basis.
Also, from a validation perspective, Agile projects should be approached more flexibly, but still within good manufacturing practices. The general guidelines for validation, whether for traditional or agile methodologies, are identical. The key is to choose the right tools to enable the validation process in a “changing environment”. For example, instead of using traditional documents, you can build documents in modules or introduce intermediate revisions (not just major versions) to allow more frequent updating. In fact, it all comes down to the methods of documenting and later approving documents before individual phases. Poor documentation, even in the case of a traditional approach, can ground a project, especially an Agile project. Apart from documents, it is of course important to look at the testing phase and, again, to see whether only parts of the system can be verified during individual sprints. To make sure that the whole system works properly as an integrated whole, you can plan for a regression test phase at the end of the project or end-to-end cross-sectional tests throughout the whole system. With good planning, such tests can be automated beforehand, which will also speed up the whole testing process.
So which methodology is best to choose for validation projects?
The best solution is to carry out the project in a hybrid methodology, for example, AgilePM, i.e. when the main phases will be conducted traditionally with all formal requirements preserved and broken down into the lowest phases, i.e. those groups of tasks that can be divided into iterations specific to the Agile methodology.
With any project, it is very important to have a good work organization, a work plan, a schedule and a flow of information so that everyone involved knows what they are responsible for and they know the project progress. This minimizes the number of mistakes that can happen. It is also important to cooperate with the ordering party and support its close involvement in the project. The exchange of information between the ordering party and the project team is then definitely better and it is certainly easier to deliver a project that is 100% compliant with the expectations in a shorter time.
Agile methodologies are not a cure for all ills, but they can sometimes speed up a project. Before starting a project, it is important to carry out an analysis of the project based on, for example, a requirement analysis, which will give an idea of how this project can be led. From the point of view of validation processes, whatever methodology is chosen, an important aspect is to involve the validator from the beginning of the process so that their comments and recommendations, as well as the work to be done throughout the project, can be taken into account. This will save everyone time and eliminate downtime, which is what not only the client but also the contractor is most concerned about.